If a connected medical device lacks the FDA’s required cybersecurity details, its submission can be refused and review can be delayed by 6 to 12 weeks.
I’d sum it up like this: FDA cybersecurity labeling is now tied to submission acceptance, patient safety, and hospital buying decisions. Since October 1, 2023, the FDA has enforced a Refuse to Accept policy for missing cyber content, and the rules now reach across 510(k), PMA, De Novo, HDE, and PDP submissions for qualifying cyber devices.
Here’s what matters most:
- The law now sets clear submission duties. Section 524B requires a vulnerability monitoring plan, cybersecurity processes, and an SBOM.
- Labeling is no longer just product paperwork. It must help hospitals and care teams set up, monitor, patch, and retire devices.
- Scope is broad. Devices with software plus internet or other network connection paths - like Wi-Fi, Bluetooth, USB, Ethernet, cellular, or cloud links - may fall under these rules.
- FDA expects specific security details. That includes interfaces, default settings, access controls, update steps, logging, residual risk, and decommissioning steps.
- Hospitals use this data before deployment. Many health systems want cybersecurity disclosures before putting devices on their networks.
- The risk is not small. One cited report says 53% of digital medical devices and internet-connected products have critical flaws, and the 2017 pacemaker recall affected nearly 500,000 devices.
A short way to think about it: if you make, buy, review, or support connected medical devices, you now need cybersecurity labeling that people can use, not just file away.
The article then walks through the legal basis, which devices count as cyber devices, what the FDA wants in labeling, where teams often miss key details, and how these expectations change across hospital systems, SaMD, cloud-connected tools, and home-use or implantable devices.
A Quick Primer on FDA's Final Guidance for Cybersecurity in Medical Devices

sbb-itb-535baee
The Regulatory Basis for FDA Cybersecurity Labeling
Two parts of the Federal Food, Drug, and Cosmetic (FD&C) Act are the legal base for FDA cybersecurity labeling and submission rules. Put simply, they spell out what manufacturers need to disclose. Those duties then show up in the exact items FDA expects to see.
FD&C Act Section 502(f) and Section 524B Requirements
Section 502(f) can turn weak cybersecurity labeling into a misbranding problem for cyber devices. If the cybersecurity information is not adequate, the device can be considered misbranded under section 502(f) [6].
Section 524B is the newer and more direct authority. It was added by Section 3305 of the Food and Drug Omnibus Reform Act of 2022 and applies to qualifying submissions made on or after March 29, 2023 [2]. Section 524B(b) calls for three core submission elements:
- A postmarket vulnerability monitoring plan that identifies and addresses vulnerabilities and exploits in a reasonable time through third-party risk management to help measure cybersecurity in healthcare
- Processes and procedures that reasonably assure device and system cybersecurity
- An SBOM that covers all software components, including commercial and open-source software [2]
Which Devices Qualify as Cyber Devices
Scope comes first. Section 524B applies only to cyber devices. Under Section 524B(c), a cyber device is a device that includes software, whether embedded or functioning as the device itself, can connect to the internet, and has characteristics that create cybersecurity risk [2].
In day-to-day use, that connectivity bar is fairly broad. FDA guidance points to wireless protocols, inductive communications, physical ports, and networked services as possible triggers [6].
| Connectivity Type | Examples |
|---|---|
| Wireless protocols | Wi-Fi, Bluetooth, Bluetooth Low Energy (BLE), cellular |
| Inductive | Magnetic inductive communications |
| Physical ports | USB, Ethernet, serial port |
| Networked services | Cloud or server connections |
The same rules also apply when an existing authorized device is changed in a way that calls for a new premarket submission [2].
Premarket Submissions and FDA Guidance That Include Labeling Expectations
Section 524B applies across the main premarket pathways: 510(k) (Traditional, Special, and Abbreviated), PMA, De Novo, HDE, and PDP submissions [5][2].
The FDA finalized its guidance, Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions, on June 27, 2025 [5]. That document lays out the documentation reviewers expect to see. It is nonbinding guidance, but it reflects FDA current thinking and shows what reviewers will be looking for in submissions [5].
In practice, manufacturers should also line up development work with a Secure Product Development Framework (SPDF) and consensus standards such as IEC 81001-5-1 [6]. MDS2 is still a common, nonrequired format for sharing security capabilities with healthcare organizations [6].
These statutory requirements are the starting point for the labeling content that follows.
What FDA Expects in Cybersecurity Labeling
Building on Section 524B, FDA expects labeling to give HDOs enough detail to deploy, secure, monitor, and retire the device safely.
Configuration, Access Control, and Interface Details
Labeling should spell out every interface the device uses, its default state, and whether it can be turned off. It should also include a network diagram that shows trust boundaries and data flows [1].
Access settings matter too. Labeling should state the TLS version, password policy, session timeout, lockout threshold, MFA, RBAC, SSO options, and any break-glass access path. If break-glass access exists, the labeling should document how it works and how audit logging tracks its use [1][3].
SBOMs, Updates, Patching, and Vulnerability Disclosure
Once the device is in use, labeling also needs to explain what software is inside it and how updates happen.
That means including a summary of software components and versions, plus a clear way for authorized users to get the full SBOM. In practice, that usually means a customer portal or a documented request workflow [1][3].
Update instructions should cover:
- How updates are delivered
- How authenticity checks are performed
- Expected downtime
- Rollback steps if something goes wrong
Labeling should also list vulnerability disclosure channels and the locations of security advisories [1][3].
Logging, Residual Risk, and Decommissioning Instructions
FDA also expects labeling to show how the device records security-related activity. It should document which events are logged, the log format, export options, and support for syslog or SIEM tools. Time sync requirements should be included too, usually through NTP, so timestamps stay reliable during an investigation [1][3].
Some risk won't disappear, and FDA wants that stated plainly. Labeling should disclose unmitigable residual risks, their clinical impact, and any compensating controls [1][3]. Decommissioning instructions should also cover data sanitization, account removal, and how to handle keys or certificates when the device is taken out of service [1][3].
How Transparency Standards Affect Manufacturers and HDOs
Using Standardized Disclosures to Support Healthcare Risk Decisions
Once FDA-required disclosures are in place, HDOs can use them to compare devices and plan deployment with a lot more confidence. Those disclosures now play a direct role in how HDOs review devices before they ever touch the network.
The catch is simple: labeling has to stay current. Vulnerabilities change. Configurations change. If the documentation stays frozen while the device keeps changing, the data stops being useful fast.
That’s why clear labeling, SBOMs, and MDS2 forms matter so much. They give procurement and biomed teams concrete data instead of vague promises.
MDS2 and JSP2 also make side-by-side reviews easier. They help HDOs spot missing controls and plan segmentation before deployment. In plain terms, these forms turn vendor claims into day-to-day security decisions, giving teams a steady way to judge device risk before it reaches the network.
Putting Labeling Data to Work with Censinet
Of course, documents alone don’t fix anything. Teams need a repeatable workflow that turns labeling data into action.
Censinet RiskOps™ helps HDOs organize labeling data across third-party and enterprise risk workflows, support benchmarking, and keep human reviewers in control. That matters during medical device deployment and monitoring, where labeling data needs to guide segmentation decisions and lifecycle management.
Common Labeling Gaps That Create Downstream Risk
When disclosures are incomplete, the problem usually shows up during deployment, not on paper.
Three labeling failures drive most of the downstream risk:
- Incomplete interface documentation that leaves out disabled-by-default or future-enabled ports
- Vague hardening guidance that gives IT teams nothing they can actually configure
- Missing end-of-support dates that leave unpatchable legacy devices on clinical networks with no replacement plan
When labeling leaves out these details, HDOs can’t segment networks, plan replacements, or apply compensating controls in a useful way.
Applying FDA Cybersecurity Labeling Expectations Across Device Types and Next Steps
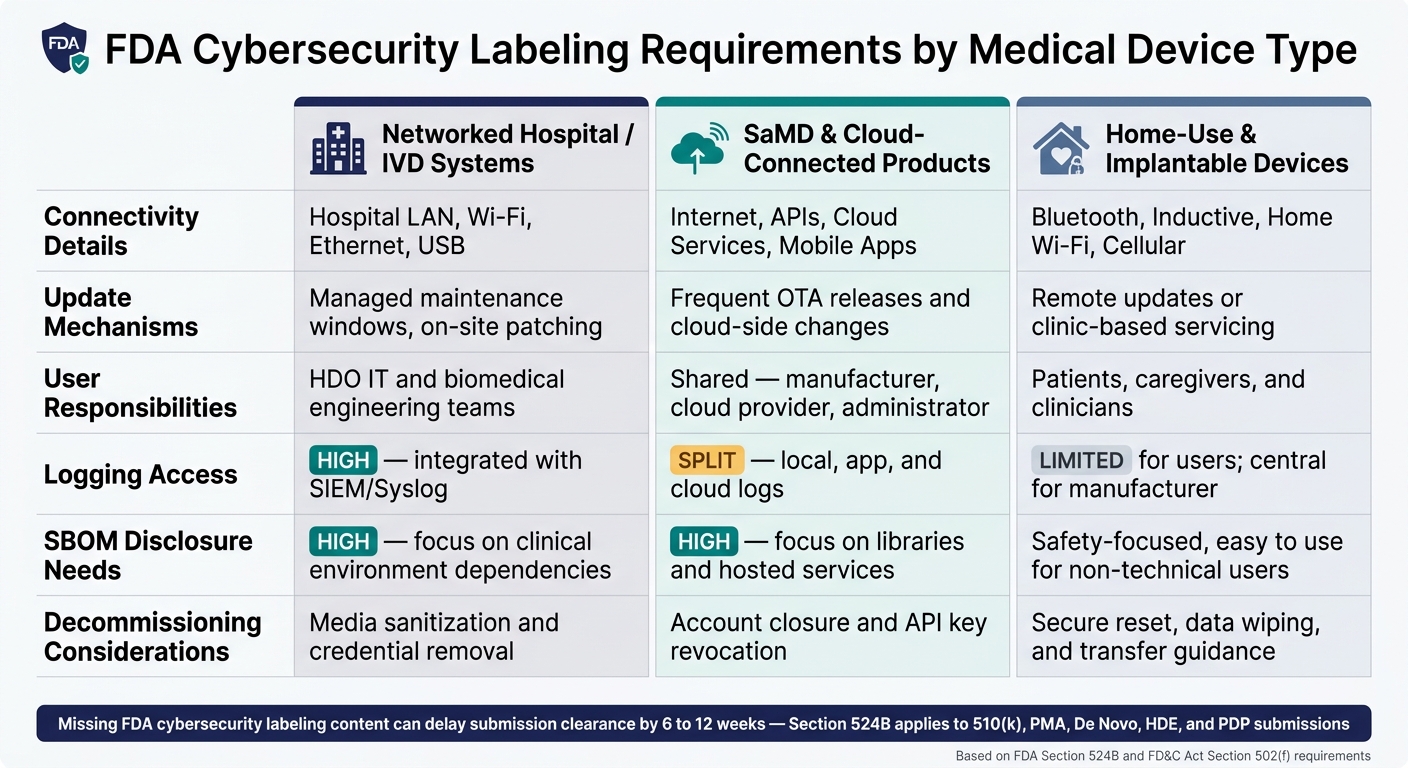
FDA Cybersecurity Labeling Requirements by Medical Device Type
How Labeling Priorities Differ by Device Category
FDA labeling should match how a device connects, how it gets updates, and who actually uses it.
The core disclosure rules stay the same across device types. But the part that needs the most attention changes based on where the device lives and who manages it.
| Device category | Connectivity details | Update mechanisms | User responsibilities | Logging access | SBOM disclosure needs | Decommissioning considerations |
|---|---|---|---|---|---|---|
| Networked hospital/IVD systems | Hospital LAN, Wi-Fi, Ethernet, USB | Managed maintenance windows, on-site patching | HDO IT and biomedical engineering teams | High; integrated with SIEM/Syslog | High; focus on clinical environment dependencies | Media sanitization and credential removal |
| SaMD and cloud-connected products | Internet, APIs, cloud services, mobile apps | Frequent OTA releases and cloud-side changes | Shared across the manufacturer, cloud provider, and administrator | Split across local, app, and cloud logs | High; focus on libraries and hosted services | Account closure and API key revocation |
| Home-use or implantable devices | Bluetooth, inductive communications, home Wi-Fi, cellular | Remote updates or clinic-based servicing | Patients, caregivers, and clinicians | Limited for users; central for manufacturer | Safety-focused and easy to use for non-technical users | Secure reset, data wiping, and transfer guidance |
Why does this matter? Because the person using the device, the way updates happen, and the party that owns the logs all shift by category.
A networked hospital system usually sits in an IT-managed setting. A home-use or implantable device is different. In that case, labeling has to work for patients and caregivers too, not only clinical teams.
For SaMD and cloud-connected products, software can change often. That means labeling needs to separate routine releases from security patches and spell out who can approve each one [4].
Key Takeaways for Manufacturers and Healthcare Organizations
FDA cybersecurity labeling now plays into premarket review. If required content is missing, manufacturers can get Additional Information requests that delay clearance by 6 to 12 weeks [1].
For manufacturers, the job doesn't stop at submission. Disclosures need updates when CVEs, supported configurations, or protocols change, including changes that happen after submission [1] [4].
For healthcare organizations, these disclosures are more than paperwork. They can guide decisions about segmentation, procurement, and lifecycle planning. Censinet RiskOps™ can help HDOs put labeling data into structured risk workflows.
FAQs
Does my device qualify as a cyber device?
A device qualifies as a cyber device under Section 524B(c) of the FD&C Act if it:
- includes software validated, installed, or authorized by the sponsor
- can connect to the internet, whether on purpose or not
- has tech features validated, installed, or authorized by the sponsor that could be open to cybersecurity threats
If you're unsure, you may contact the FDA for clarification.
What happens if cybersecurity labeling is incomplete?
If a device’s cybersecurity labeling is incomplete, the FDA may classify it as misbranded under section 502(f) of the Federal Food, Drug, and Cosmetic Act. That’s not a minor paperwork issue. It can lead to serious regulatory action, including market removal, recalls, and even possible criminal charges.
The risk starts early, too. In premarket submissions, weak or incomplete cybersecurity documentation can trigger a Refuse to Accept decision, which can slow down market entry. On top of that, poor labeling can make it harder for healthcare organizations to manage risk in practice.
How should manufacturers keep labeling current after clearance?
Manufacturers should keep labeling current by building cybersecurity into the full device lifecycle as part of a Total Product Life Cycle approach.
That means having clear processes to monitor, identify, and fix vulnerabilities over time, including Coordinated Vulnerability Disclosure procedures.
They should also use tools like a Predetermined Change Control Plan for approved security updates, update Software Bill of Materials documentation with each release, and clearly communicate end-of-support dates.
